
We use multidisciplinary approaches ranging from human molecular genetics, molecular and cellular biology, biochemistry to developmental biology and mouse genetics to understand the physiology and pathophysiology of polycystins and polycystic kidney disease (PKD) which has led us to the studies of primary cilia.
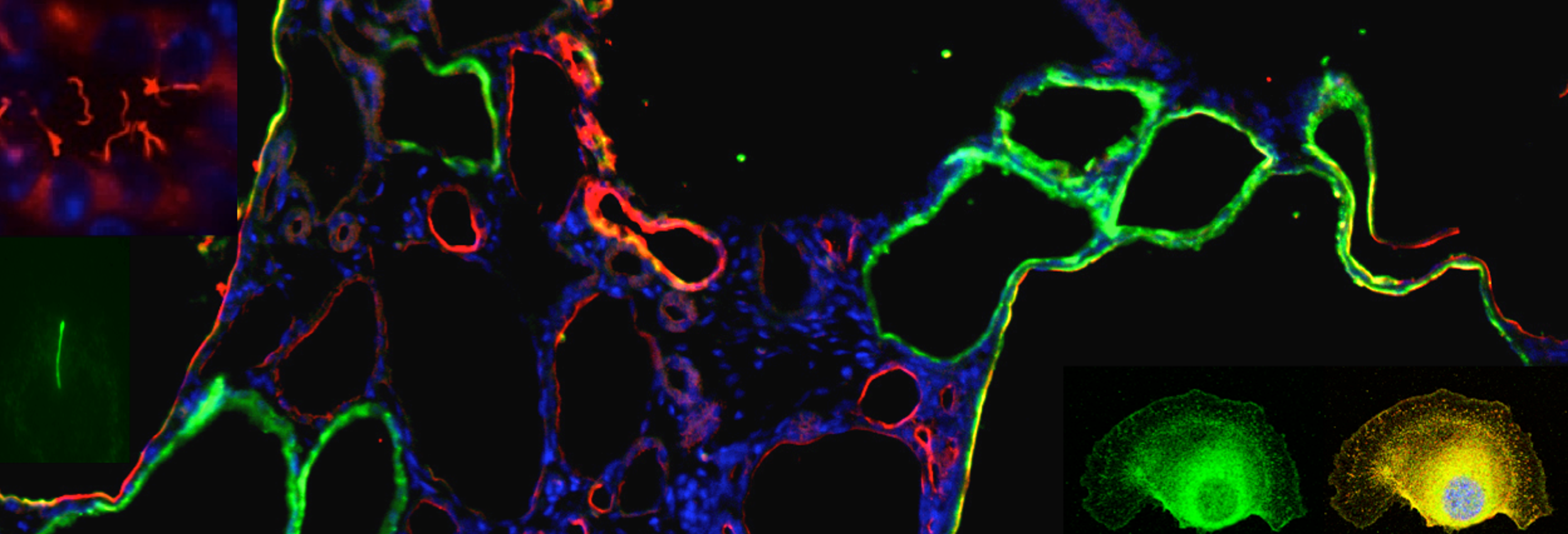
Autosomal dominant polycystic kidney disease (ADPKD) is the most common monogenetic disorder in humans with an estimated prevalence of 1:1000 affecting ~12.5 million people worldwide. The main characteristic of ADPKD is the progressive development of epithelial lined cysts in the kidney, which ultimately cause loss of renal function. The mechanisms of cyst development remain unclear. The disease is caused by mutations in PKD1 and PKD2 genes which encode polycystin-1 and -2, respectively. We have shown, by gene targeting experiments, a critical role of polycystin-1 in kidney and pancreas development. We demonstrated that polycystin-L and -2 are calcium-permeable cation channels, and that polycystin-1 acts as an atypical G-protein coupled receptor whose functionality is coupled with polycystin-2.
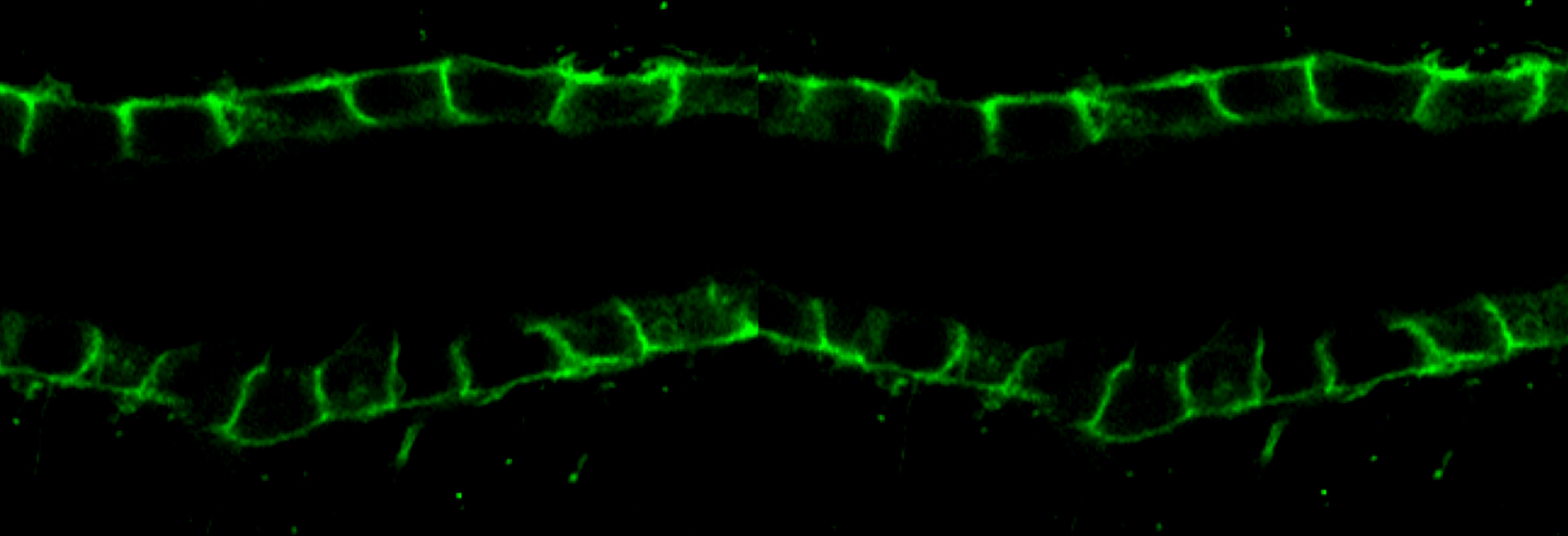
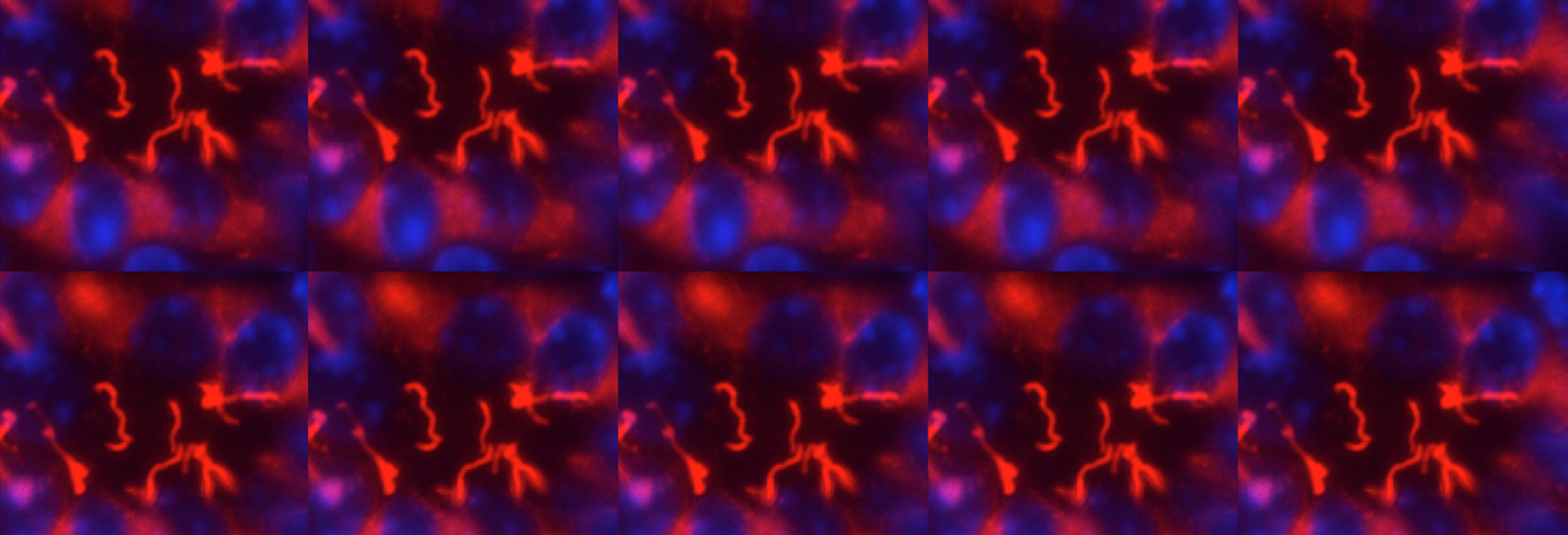
During development, tubules can arise from cells in many different starting conditions and configurations, and their lumen size is strictly controlled. Our premise is that these diverse pathways converge on a smaller number of common downstream mechanisms that directly control the organization of epithelial cells into tubules. These mechanisms are poorly understood and sit at the crossroads of developmental and cell biology. Cyst formation in PKD, by definition, is characterized by the dilation of tubules -the loss of normal size control of tubular lumen. Cell biological studies of polycystic kidney have shown that the loss of tube lumen size control is accompanied by increased cell proliferation, decreased cell differentiation and cell survival. Using cells cultured from mouse models defective in polycystin-1, we have recently demonstrated, for the first time, that polycystin-1 and -2 mediate mechanosensation in the primary cilium of kidney epithelium. Primary cilium of kidney epithelium has been previously proposed to be a mechanosensor with unknown molecular composition. Our data suggest polycystin-1 and -2 functions as a mechanosensitve receptor-channel complex that controls normal tubular morphogenesis. Our recent discovery show that the orientation of cell division in postnatal tubules of Pkd1 knockout mice is randomized, in striking contrast to the cells in normal kidneys which divide in an orientation that is in parallel to the tubular axis.
One of our discoveries is that the ADPKD protein polycystin-2 is in the same protein complex as FPC, the protein encoded by the gene PKHD1 whose mutation causes a recessive form of PKD in infants and children. Our current focus is to understand the downstream signaling events of the ADPKD and ARPKD proteins. Particularly how these proteins are involved in cell differentiation and survival, in spindle bipolarity, in cell cycle progression, cell division, cilia and centromsome biology, and the signaling pathways downstream from shear stress induced Ca2+ signal. We are also interested in collaborations with other groups to study the role of polycystins in other organs and cell types as polycystins are expressed in multiple tissues with diverse functions. For example, polycystin-2 controls the left-right body axis and fertility. We are also using multidisciplinary approaches to identify the functions of four polycystin-like molecules identified in our lab.
Techniques used include mammalian cell culture (including primary cell culture from tissues), immunocytochemistry, transient and stable transfection, in vitro 3D tubulogenesis assays, live cell imaging, and in vivo tissue- and time- specific gene targeting, molecular cloning, yeast-two-hybrid assay, Western analysis, co-immunoprecipitation, Ca2+ imaging.